From Carl R. Woese Institute for Genomic Biology - University of Illinois Urbana-Champaign
Jump to navigation
Jump to search
Quick Links[edit]
Description[edit]
Biocluster is the High Performance Computing (HPC) resource for the Carl R Wose Institute for Genomic Biology (IGB) at the University of Illinois at Urbana-Champaign (UIUC). Containing 2824 cores and over 27.7 TB of RAM, Biocluster has a mix of various RAM and CPU configurations on nodes to best serve the various computation needs of the IGB and the Bioinformatics community at UIUC. For storage, Biocluster has 2619 TB of storage on its Ceph filesystem for reliable high speed data transfers within the cluster. Networking in Biocluster is either 1 or 10 Gigibit ethernet depending on the class of node and its data transfer needs.
Cluster Specifications[edit]
| Queue Name
|
Nodes
|
Cores (CPUs) per Node
|
Memory
|
Networking
|
Scratch Space /scatch
|
GPUs
|
|---|
| normal (default)
|
5 Supermicro SYS-2049U-TR4
|
72 Intel Xeon Gold 6150 @2.7 GHz
|
1.2TB
|
40GB Ethernet
|
7TB SSD
|
|
| himemeory
|
2 Supermicro
|
28 Intel Xeon E5-2690 v4 @ 2.6GHz
|
768GB
|
10GB Ethernet
|
4TB SSD
|
|
| lowmem
|
8 Supermicro SYS-F618R2-RTN+
|
12 Intel Xeon E5-2603 v4 @ 1.70Ghz
|
64GB RAM
|
10GB Ethernet
|
192GB
|
|
| gpu
|
1 Supermicro
|
28 Intel Xeon E5-2680 @ 2.4Ghz
|
128GB
|
1GB Ethernet
|
1TB
|
4 NVIDIA GeForce GTX 1080 Ti
|
| classroom
|
22 Dell Poweredge PE1950
|
8 Intel Xeon E5440 @ 2.83GHz
|
16GB
|
1GB Ethernet
|
500GB
|
|
Storage[edit]
Information[edit]
- The storage system is a CEPH filesystem with 2.4 Petabytes of disk space. This data is NOT backed up.
- The data is spread across 15 CEPH storage nodes.
| Cost (Per Terabyte Per Month)
|
|---|
| $10
|
Queue Costs[edit]
The cost for each job is dependent on which queue it is submitted to. Listed below are the different queues on the cluster with their cost.
Usage is charge by the second. The CPU cost and memory cost are compared and the largest is what is billed.
| Queue Name
|
CPU Cost ($ per CPU per day)
|
Memory Cost ($ per GB per day)
|
GPU Cost ($ per GPU per day)
|
|---|
| normal (default)
|
$1.00
|
$0.07
|
NA
|
| himemory
|
$2.00
|
$0.07
|
NA
|
| lowmem
|
$0.50
|
$0.10
|
NA
|
| GPU
|
$2.00
|
$0.44
|
$2.00
|
}
Gaining Access[edit]
Cluster Rules[edit]
- Running jobs on the head node or login nodes are strictly prohibited. Running jobs on the head node could cause the entire cluster to crash and affect everyone's jobs on the cluster. Any program found to be running on the headnode will be stopped immediately and your account could be locked. You can start an interactive session to login to a node to manual run programs.
- Installing Software Please email help@igb.illinois.edu for any software requests. Compiled software will be installed in /home/apps. If its a standard RedHat package (rpm), it will be installed in their default locations on the nodes.
- Creating or Moving over Programs: Programs you create or move to the cluster should be first tested by you outside the cluster for stability. Once your program is stable, then it can be moved over to the cluster for use. Unstable programs that cause problems with the cluster can result in your account being locked. Programs should only be added by CNRG personnel and not compiled in your home directory.
- Reserving Memory: SLURM allows the user to specify the amount of memory they want their program to use.. If your job tries to use more memory than you have reserved, the job will run out of memory and die. Make sure to specify the correct amount of memory.
- Reserving Nodes and Processors: For each job, you must reserve the correct number of nodes and processors. By default you are reserved 1 processor on 1 node. If you are running a multiple processor job or a MPI job you need to reserve the appropriate amount. If you do not reserve the correct amount, the cluster will confine your job to that limit, increasing its runtime.
How To Log Into The Cluster[edit]
- You will need to use an SSH client to connect.
On Windows[edit]
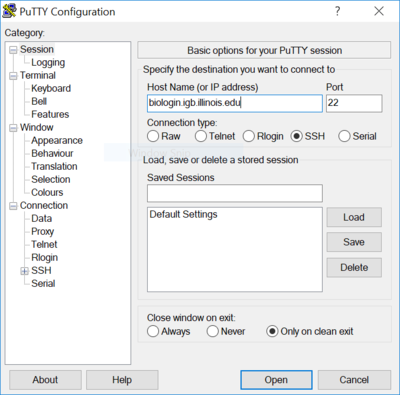
- Hit Open and login using your IGB account credentials.
On Mac OS X[edit]
- Simply open the terminal under Go >> Utilities >> Terminal
- Type in ssh username@biologin.igb.illinois.edu where username is your NetID.
- Hit the Enter key and type in your IGB password.
How To Submit A Cluster Job[edit]
Create a Job Script[edit]
- You must first create a SLURM job script file in order to tell SLURM how and what to execute on the nodes.
- Type the following into a text editor and save the file test.sh ( Linux text editing )
#!/bin/bash
#SBATCH -p normal
#SBATCH --mem=1g
#SBATCH -N 1
#SBATCH -n 1
sleep 20
echo "Test Script"
- You just created a simple SLURM Job Script.
- To submit the script to the cluster, you will use the sbatch command.
sbatch test.sh
- Line by line explanation
- #!/bin/bash - tells linux this is a bash program and it should use a bash interpreter to execute it.
- #SBATCH - are SLURM parameters, for explanations of these please scroll down to SLURM Parameters Explanations section.
- sleep 20 - Sleep 20 seconds (only used to simulate processing time for this example)
- echo "Test Script" - Output some text to the screen when job completes ( simulate output for this example)
- For example if you would like to run a blast job you may simply replace the last two line with the following
module load BLAST
blastall -p blastn -d nt -i input.fasta -e 10 -o output.result -v 10 -b 5 -a 5
- Note: the module commands are explained under the Environment Modules section.
SLURM Parameters Explanations:[edit]
- To view all possible parameters
| Command
|
Description
|
|---|
| #SBATCH -p PARTITION
|
Run the job on a specific queue/partition. This defaults to the "normal" queue
| | #SBATCH -D /tmp/working_dir
|
Run the script from the /tmp/working_dir directory. This defaults to the current directory you are in.
| | #SBATCH -J ExampleJobName
|
Name of the job will be ExampleJobName
| | #SBATCH -e /path/to/errorfile
|
Split off the error stream to this file. By default output and error streams are placed in the same file.
| | #SBATCH -o /path/to/ouputfile
|
Split off the output stream to this file. By default output and error streams are placed in the same file.
| | #SBATCH --mail-user username@illinois.edu
|
Send an e-mail to specified email to receive job information.
| | #SBATCH --mail-type BEGIN, END, FAIL
|
Specifies when to send a message to email. You can select multiple of these with a comma separated list. Many other options exist.
| | #SBATCH -N X
|
Reserve X number of nodes.
| | #SBATCH -n X
|
Reserve X number of cpus.
| | #SBATCH --mem=XG
|
Reserve X gigabytes of RAM for the job.
| | #SBATCH --gres=gpu:X
|
Reserve X NVIDIA GPUs. (Only on GPU queues)
|
Create a Job Array Script[edit]
Making a new copy of the script and then submitting each one for every input data file is time consuming. An alternative is to make a job array using the -t option in your submission script. The ---aray option allows many copies of the same script to be queued all at once. You can use the $SLURM_ARRAY_TASK_ID to differentiate between the different jobs in the array. A brief example on how to do this is available at Job Arrays
Start An Interactive Session[edit]
- Use the ```srun``` commsnf if you would like to run a job interactively.
srun --pty /bin/bash
- This will automatically reserve you a slot on one of the compute nodes and will start a terminal session on it.
- Closing your terminal window will also kill your processes running in your interactive srun session, therefore it's better to submit large jobs via non-interactive sbatch.
X11 Graphical Applications[edit]
- To run an application with a user interface you will need to setup an Xserver on your computer Xserver Setup
- Then add the ```--x11``` parameter to your srun command
srun --x11 --pty /bin/bash
View/Delete Submitted Jobs[edit]
Viewing Job Status[edit]
- To get a simple view of your current running jobs you may type:
squeue -u userid
- This command brings up a list of your current running jobs.
- The first number represents the job's ID number.
- Jobs may have different status flags:
- R = job is currently running
- For more detailed view type:
squeue -l
- This will return a list of all nodes, their slot availability, and your current jobs.
List Queues[edit]
sinfo This will show all queues as well as which nodes in those queues are fully used (alloc), partially used (mix), unused (idle), or unavailable (down).
List All Jobs on Cluster With Nodes[edit]
squeue
Deleting Jobs[edit]
- Note: You can only delete jobs which are owned by you.
- To delete a job by job-ID number:
- You will need to use scancel, for example to delete a job with ID number 5523 you would type:
scancel 5523
scancel -u userid
Troubleshooting job errors[edit]
- To view job errors in case job status shows Eqw or any other error in the status column use qstat -j, for example if job # 23451 failed you would type:
scontrol show job 23451
Applications[edit]
Application Lists[edit]
Application Installation[edit]
Environment Modules[edit]
- The Biocluster uses the Lmod modules package to manage the software that is installed. You can read more about Lmod at https://lmod.readthedocs.io/en/latest/
- To use an application, you need to use the module command to load the settings for an application
- To load a particular environment for example QIIME/1.9.1, simply run this command:
module load QIIME/1.9.1
- If you would like to simply load the latest version, run the the command without the /1.9.1 (version number):
module load QIIME
- To view which environments you have loaded simply run module list:
bash-4.1$ module list
Currently Loaded Modules:
1) BLAST/2.2.26-Linux_x86_64 2) QIIME/1.9.1
- When submitting a job using a sbatch script you will have to add the module load qiime/1.5.0 line before running qiime in the script.
- To unload a module simply run module unload:
module unload QIIME
module purge
Mirror Service - Genomic Databases[edit]
Transferring data files[edit]
Transferring using SFTP/SCP[edit]
Using WinSCP[edit]
- Download WinSCP installation package from http://winscp.net/eng/download.php#download2 and install it.
- Once installed Run WinSCP >> enter biologin.igb.illinois.edu for the Host name >> Enter your IGB user name and password and click Login
- Once you hit "Login, you should be connected to your Biocluster home folder, as shown below.
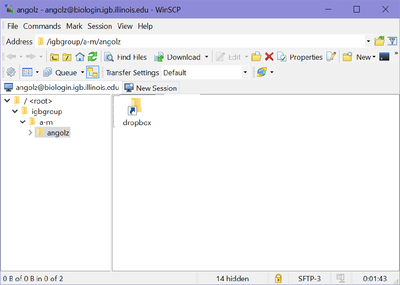
- From here you should be able to download or transfer your files.
Using CyberDuck[edit]
- To download cyberduck go to http://cyberduck.c and click on the large Zip icon to download.
- Once cyberduck is installed on OSX you may start the program.
- Click on Open Connection.
- From the drop down menu at the top of the opopup window select SFTP(SSH File Transfer Protocol)
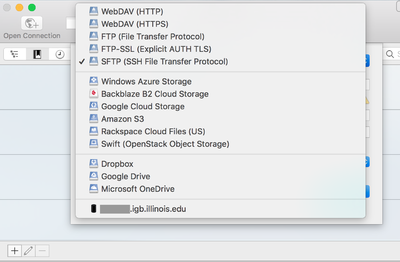
- Now in the Server: input box enter biologin.igb.illinois.edu and for Username and password enter your IGB credentials.
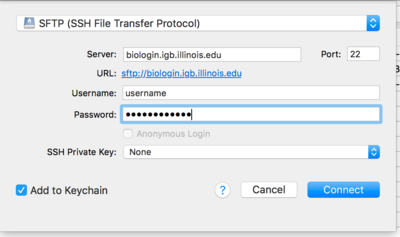
- Click Connect.
- You may now download or transfer your files.
Transferring using Globus[edit]
- The biocluster has a Globus endpoint setup. Then end point name is igb#biocluster.igb.illinois.edu
- Globus allows the transferring of very large files reliably.
- A guide on how to use Globus is here
Transferring from Biotech FTP Server[edit]
- One option to transfer data from the Biotech FTP server is to use a program called sftp.
- It can download 1 file or an entire directory.
- Replace USERNAME with the username provided by the Biotech Center.
The below example will download the file test_file.tar.gz
sftp USERNAME@ftp.biotech.illinois.edu:test_file.tar.gz
- If you want to download an entire directory, you need to have the -r parameter set. This will recursively go through an entire directory and download it into the current directory. Make sure to have the final dot at the end of the command
sftp -r USERNAME@ftp.biotech.illinois.edu:test_dir/ .
References[edit]
|




